
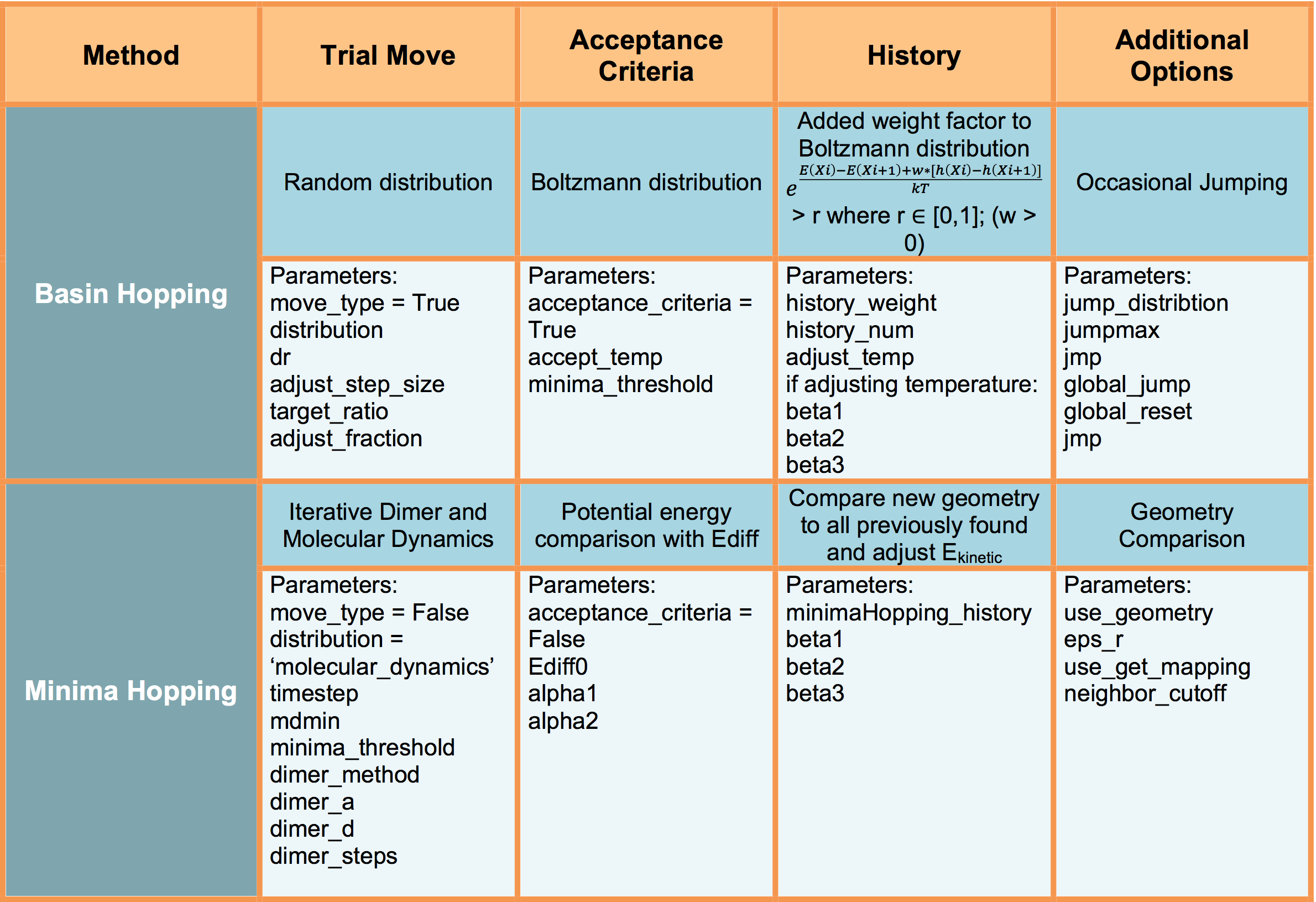
TSASE contains a software package which combines features from multiple global optimization (GO) methods including Basin-Hopping 1 and Minimia Hopping 2. In this software package, you can select a trial move, the acceptance criteria, utilize history, and take an optional jump after the acceptance criteria.
Below outlines the features of this combined GO software:
class
tsase.optimizer.Hopping (self, atoms, temperature=500, optimizer=SDLBFGS, fmax=0.01, dr=0.45, adjust_cm=True, mss=0.1, minenergy=None, move_type=True,distribution=’uniform’, adjust_step_size=10, target_ratio=0.5, adjust_fraction=0.05, significant_structure=True, pushapart=0.1, jumpmax=None, jmp=7, minima_threshold = 4, global_jump=None, global_reset=False, jump_distribution=’uniform’, dimer_method=True,dimer_a=0.001, dimer_d =0.01, dimer_steps=14, timestep=0.1, mdmin=2, history_weight=0.0, history_num=0, adjust_temp=False, accept_temp=8000, acceptance_criteria=True, minimaHopping_history=True, beta1=1.04, beta2=1.04, beta3=1.0/1.04, Ediff0=0.5, alpha1=0.98, alpha2=1./0.98, use_geometry=False, eps_r=0.1, use_get_mapping=True, neighbor_cutoff=2.3)
General GO parameters
atoms : atoms object defining the PES
temperature : temperature in Kelvin
optimizer : local optimizer (QuasiNewton, FIRE, SDLBFGS)
fmax : magnitude of the L2 norm used as the convergence criteria for local optimization
adjust_cm : fix the center of mass
mss : maximum stepsize for the local optimization
minenergy : the GO algorithm stops when a configuration is found with a lower potential energy than this value
pushapart : push atoms apart until all atoms are no closer than this distance
keep_minima_arrays : True: creates global_minima and local_minima arrays of length maximum number of Monte Carlo steps; this will result in extra zeros at the end of the array if the run finishes before the maximum number of Monte Carlo steps are taken
minima_threshold : the number of digits after the decimal point to round potential energies
check_bonds : True: check that all atoms in the system are still connected after the trial move
max_localops : True: quit the run when the number of local optimizations > steps (parameter of run())
Selecting Move Type
move_type : type of trial move (True = BH; False = MH); If move_type is set to False, you will perform a trial move specified by your selected distribution (see flag below) until you have escaped the current minimum. If the flag minimaHopping_history is set to True you will perform trial moves until you have both escaped the minima and found a state that has never been previously visited.
distribution : the distribution used for the displacement of each atom
default: uniform
options: ‘gaussian’: the parameter dr serves as the standard deviation of the gaussian distribution
‘uniform’: a uniform random number is selected in the interval [-dr,dr]
‘linear’: a uniform random number is selected in the interval [-dr*d,dr*d] where d is the distance from the geometric center of a cluster 3
‘quadratic’: a uniform random number is selected in the interval [-dr*d*d,dr*d*d] where d is the distance from the geometric center of a cluster 3
‘molecular_dynamics’: Molecular dynamics move type
Random Move Parameters
dr : maximum displacement in each degree of freedom for Monte Carlo trial moves
adjust_step_size: adjust the step size after this many Monte Carlo steps so that a target_ratio of steps are accepted.
Set to None and step size will not be adjusted. Any positive integer will turn on this feature. Default: 10
target_ratio: specified ratio of accepted steps; Default: 0.5
adjust_fraction: the fraction by which to change the step size in order to meet the target acceptance ratio;
default: 0.05
significant_structure : displace from the optimized structures after each acceptance 4
Dynamic Move Parameters
timestep : timestep in fs
mdmin : number of times the dynamics need to pass a minima to stop the MD simulation
dimer_method : Use an iterative dimer method before MD (True or False) 6
dimer_a : dimer ajustment parameter; scalar for forces in optimization 6
dimer_d : dimer adjustment parameter; distance between two images in the dimer 6
dimer_steps : dimer adjustment parameter; number of dimer iterations 6
Occasional Jumping
jump_distribution : options are the same as the distribution flag (‘uniform’, ‘molecular dynamics’, etc.)
jumpmax : after this number of consecutive rejected Monte Carlo or Minima Hopping moves, accept the following jmp moves. This allows for a more global search of the potential energy surface. 5
jmp : number of consecutive accepted moves when occassionally jumping. 5
global_reset : reset history if an occasional jump is taken; default is False
global_jump : After visiting a state for global_jump number of times, accept the following jmp moves of jump_distribution. Set to None to turn off.
Selecting Acceptance Criteria
acceptance_criteria : This flag specifies which acceptance criteria to use. True for BH or False for MH
BH Acceptance Parameters
accept_temp : seperate temperature to use for BH acceptance
adjust_temp : dynamically adjust the temperature in BH acceptance (True or False)
history_weight : weight factor for BH history, set to 0.0 to turn off history 7
history_num : limit of previously accepted minima to keep track of for BH history, set to 0 to keep track of all accepted minima
Minima Hopping Acceptance Parameters
beta1 : 1.04, temperature adjustment parameter
beta2 : 1.04, temperature adjustment parameter
beta3 : 1./1.04, temperature adjustment parameter
Ediff0 : 0.5 eV, initial energy acceptance threshold
alpha1 : 0.98, energy threshold adjustment parameter
alpha2 : 1./0.98, energy threshold adjustment parameter
minimaHopping_history : set to True if using MH history components (never accept a previously visited state) otherwise set to False
Geometry Comparision Parameters
use_geometry : compare geometries of states (True or False) when false only potential energies are compared
eps_r : positional difference to consider atoms in the same location
use_get_mapping : which geometry comparision method from atoms_operator.py to use (True for get_mapping or False for rot_match)
neighbor_cutoff : parameter for get_mapping
Below is an example script of how to use this optimizer:
from tsase.optimize.minima_basin2 import Hopping lj = tsase.calculators.lj(cutoff=35.0) system = tsase.io.read_con('lj38-cluster.con') system.set_calculator(lj) opt = Hopping(atoms=system, minenergy=-173.918427) opt(10000)Below is an example script of how to use the combined GO algorithm to run standard MH:
from tsase.io import read_con from tsase.calculators import lj from tsase.optimize.minima_basin2 import Hopping lj = lj(cutoff=35.0) system = read_con('lj38-cluster.con') system.set_calculator(lj) opt = Hopping(atoms=system, temperature=500, minenergy=-173.918427, move_type = False, distribution='molecular_dynamics', jumpmax=None, global_jump=None, global_reset=False, dimer_a=0.001, dimer_d=0.01, dimer_steps=20, timestep=0.1, mdmin=2, acceptance_criteria=False, minimaHopping_history=True, beta1=1.04, beta2=1.04, beta3=1.0/1.04, Ediff0=0.5, alpha1=0.98, alpha2=1./0.98, use_geometry=True, keep_minima_arrays=False) opt.run(10000, maxtemp=50000)Below is an example script of how to use the combined GO algorithm to run standard BH:
from tsase.io import read_con from tsase.calculators import lj from tsase.optimize.minima_basin2 import Hopping lj = lj(cutoff=35.0) system = read_con('lj38-cluster.con') system.set_calculator(lj) opt = Hopping(atoms=system, dr=0.45, minenergy=-173.918427, move_type = True, distribution='uniform', adjust_step_size=10, target_ratio=0.5, jumpmax=None, global_jump=None, global_reset=False, history_weight=0.0, history_num=0, adjust_temp=False, accept_temp = 8000, acceptance_criteria=True, use_geometry=True, keep_minima_arrays=False) opt.run(10000)
Below outlines the features of the basin hoppping implementation. All flags are equivalent to the description above except for those defined below.
class
tsase.optimizer.BasinHopping (self, atoms, temperature=100 * kB, optimizer=SDLBFGS, fmax=0.1,dr=0.1, active_ratio=1.0, adjust_cm=True, mss=0.2, minenergy=None, distribution=’uniform’, significant_structure = False, pushapart = 0.4,jumpmax=15,adjust_step_size=None, target_ratio = 0.5, adjust_fraction = 0.05)temperature : temperature in kT
from tsase.optimize.basin import BasinHopping
lj = tsase.calculators.lj(cutoff=35.0)
system = tsase.io.read_con('lj38-cluster.con')
system.set_calculator(lj)
opt = MinimaHopping(atoms=system, minenergy=-173.918427)
opt(10000)
Below outlines the features of the minima hoppping implementation. All flags are equivalent to the description in the combined GO method.
class
tsase.optimizer.MinimaHopping (self, atoms, T0, beta1, beta2, beta3, Ediff0, alpha1, alpha2, mdmin, logfile, minima_threshold, timestep, optimizer, minima_traj, fmax, dimer_a, dimer_d, dimer_steps)
Below is an example script of how to use this optimizer:
from tsase.optimize.minimahopping import MinimaHopping lj = tsase.calculators.lj(cutoff=35.0) system = tsase.io.read_con('lj38-cluster.con') system.set_calculator(lj) opt = MinimaHopping(atoms=system) opt(totalsteps=10000, maxtemp=200000, minEnergy=-173.918427)
References
Wales, J. P. K. Doye, “Global Optimization by Basin-Hopping and the Lowest Energy Structures of Lennard-Jones Clusters Containing up to 110 Atoms”, J. Phys. Chem. 101,5111-5116 (1997).
Goedecker, “Minima hopping: An efficient search method for the global minimum of the potential energy surface of complex molecular systems”, J. Chem. Phys. 120, 9911 (2004).
Kim, S. K. Choi, and H. M. Lee. “New algorithm in the basin hopping Monte Carlo to find the global minimum structure of unary and binary metallic nanoclusters.” J. Chem. Phys., 144702 (2008).
White, and H. R. Mayne. “An investigation of two approaches to basin hopping minimization for atomic and molecular clusters.” Chemical Physics Letters, 463-468 (1998).
Iwamatsu, and Y. Okabe. “Basin hopping with occasional jumping.” Chem. Phys. Lett., 396-400 (2004).
Shonborn, S. Goedecker, S. Roy, and A. Oganov, “The performance of minima hopping and evolutionary algorithms for cluster structure prediction”, J. Chem. Phys. 130, 144108 (2009).
G Rossi and R Ferrando, “Searching for low-energy structures of nanoparticles: a comparison of different methods and algorithms” J. Phys.: Condens. Matter 21, 084208 (2009).