

Unraveling the Catalytic Nature and Factors Affecting Reaction Rate Under Reaction Condition
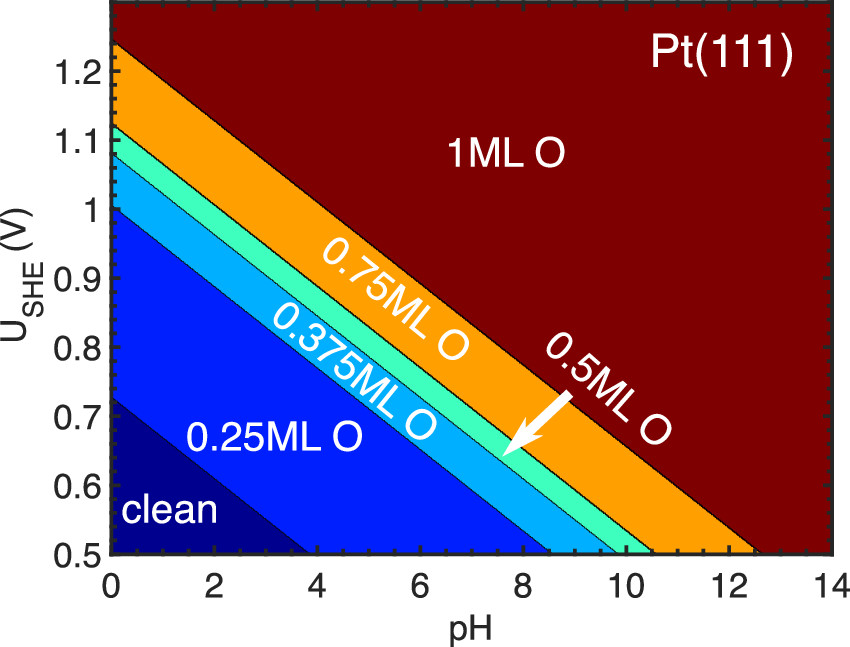
Pt dissolution under potential cycling has been identified as the dominant process that causes cathode losses in proton-exchange membrane fuel cells. In recent years, significant insights on the Pt dissolution process have been obtained from in situ Pt dissolution detection enabled by voltammetry coupled to inductively coupled plasma mass spectrometry. Despite extensive experimental research, theoretical studies continue to lag in the understanding of the atomic-scale mechanism of the Pt dissolution process due to the complicated subprocesses involved, including Pt oxidation, surface reconstruction, Pt oxide reduction, chemical corrosion, etc. Here, we employ global optimization and constant-potential density functional theory to simulate the complete process of Pt dissolution. We show that a two-dimensional Pt surface oxide consisting of interconnected square planar PtO4 units forms at applied potentials higher than 1.1 V RHE. The structural signatures and oxidation states of the Pt surface oxide are close to that of bulk Pt3O4 oxide. The PtO4 units can be reduced to [PtOH(H2O)3]+ in the cathodic scan and dissolve into the electrolyte. An anodic Pt dissolution mechanism is also proposed. Our work provides a fundamental understanding of Pt dissolution under potential cycling, which is needed for the rational design of durable Pt-based cathodes for fuel cells.
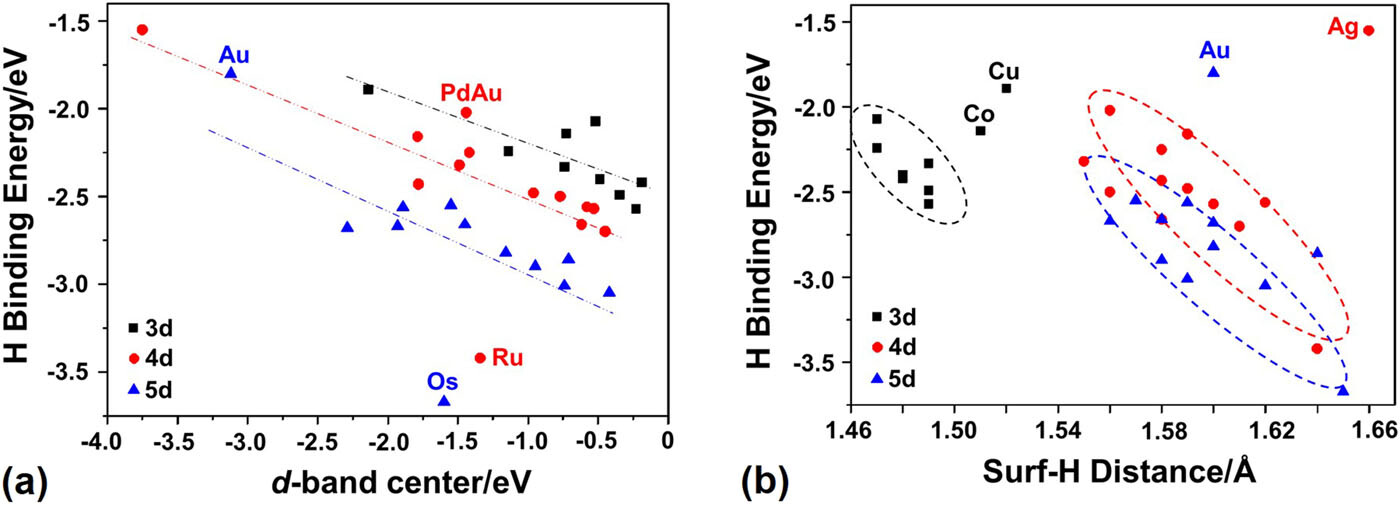
The d-band model has proven to be effective for understanding trends in the chemisorption of various adsorbates on transition metal surfaces. However, hydrogen adsorption at the atop site of transition metals and their bimetallic alloy surfaces do not always correlate well with the d-band center of the adsorption site. Additionally, the d-band model cannot explain the disappearance of the local minima for H adsorption at the hollow site on the potential energy surface of 5d single-atom element doped Au and Ag(111) surfaces. Here, we use a simple model with factors, including the d-band center, filling of the d-band, renormalized adsorbate states, coupling matrix elements, and surface–adsorbate bond lengths, to correlate with the density functional theory calculated H binding energies on both mono- and bimetallic (111) surfaces. Our results suggest that H adsorption at metal-atop sites is determined by all these factors, not only by the d-band center. The strong adsorption of H at the atop sites of 5d metal surfaces can be explained by their lower repulsive contribution.
Ceria (CeO2) has long been recognized as a critical material in using CH4 as a fuel. Many successful instances of direct-methane SOFCs have been reported with metal/oxide composite anodes, including ceria as the main component. These successes arose due to a high resistance to carbon deposition and an excellent reactivity toward hydrocarbon oxidation when using this material, presumably stemming from its inherent ability to store and release oxygen. Ceria also transports oxygen ions more rapidly than the state-of-the-art solid electrolyte, yttria-stabilized zirconia (YSZ), yet retains an electron conductivity that easily exceeds that of oxygen ions in a reducing atmosphere. To the best of our knowledge, despite numerous research efforts, the mechanism of the direct electrochemical oxidation of CH4 at the ceria/gas interface has not been clarified. Therefore, there is no effective solution to promote CH4 electrochemical oxidation, which is known to be more sluggish than that associated with H2. To solve this challenge, we investigated the direct CH4 electro-oxidation reaction at the gas/ceria electrochemical interfaces. Moreover our results revealed that the H2O formation step limits the overall reaction rate in CH4 electro-oxidation process rather than C-H cleavage or CO2 formation. Under similar reaction pathways sharing the RDS in H2 and CH4 fuels, a direct comparison of surface OH concentration clarified that how much SDC surface can be reduced is one major factor determining the performance of ceria-based anode. Our conclusion thus far suggests potential strategies to enhance the surface activity of SDC electrodes when directly utilizing CH4 as a fuel in SOFCs.
Identification of Active Sites on Surface
A better theoretical understanding on the nature of the active sites would help further optimization for target reaction. Although quantum mechanical calculations have been widely employed to elucidate the active sites over various catalysts, these calculations are typically done assuming constant-charge conditions rather than the experimentally relevant constant-potential conditions. As one of the example for theoretical approach of active sites identification, we employ the double-reference method to simulate the energetics of the oxygen reduction reaction (ORR) over pure and nitrogen-doped carbon materials under constant-potential conditions. the N-doped carbon materials are promising metal-free catalysts for the electrochemical ORR. We demonstrate that constant-potential calculations enable more accurate theoretical predictions, comparing well with existing experiments.
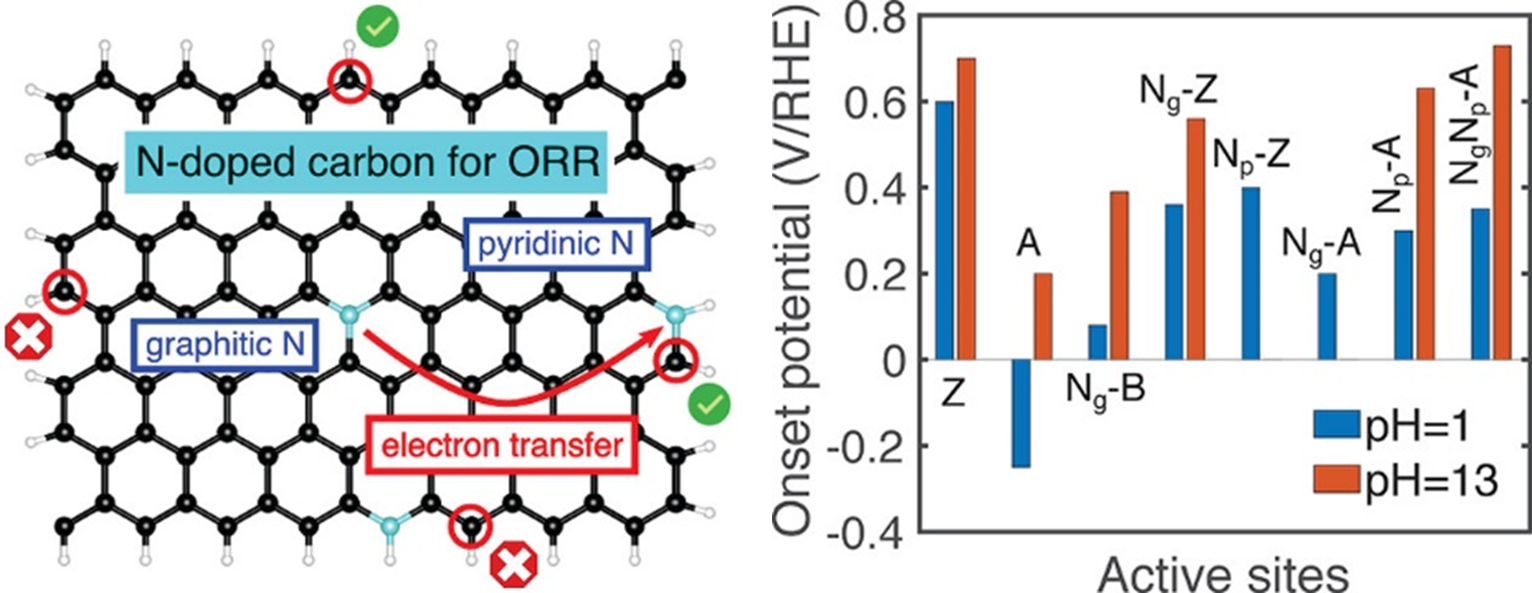
Our key findings are (1) the zigzag edge of pure graphite is highly active for ORR, (2) the pyridinic N-doped armchair edge is highly active for ORR in alkaline media but not in acid, and (3) graphitic N can donate electrons to pyridinic N to enhance the ORR activity. These fundamental insights provide guidelines for the design of better carbon-based ORR catalysts.
Understanding Fischer-Tropsch Chemistry at Surfaces
The chemistry of the Fischer-Tropsch (FT) reaction is of particular interest due to the ability to convert syngas into various sized hydrocarbons (CxHyOz). Most FT catalysts produce hydrocarbon products based on the Anderson-Schultz-Flory distribution. A few catalysts have been shown to break this distribution, which opens the door for wider tailorability of products. Of these catalysts, a number utilize promoters such as Mn, Mg, Na, and K to enhance catalytic performance.
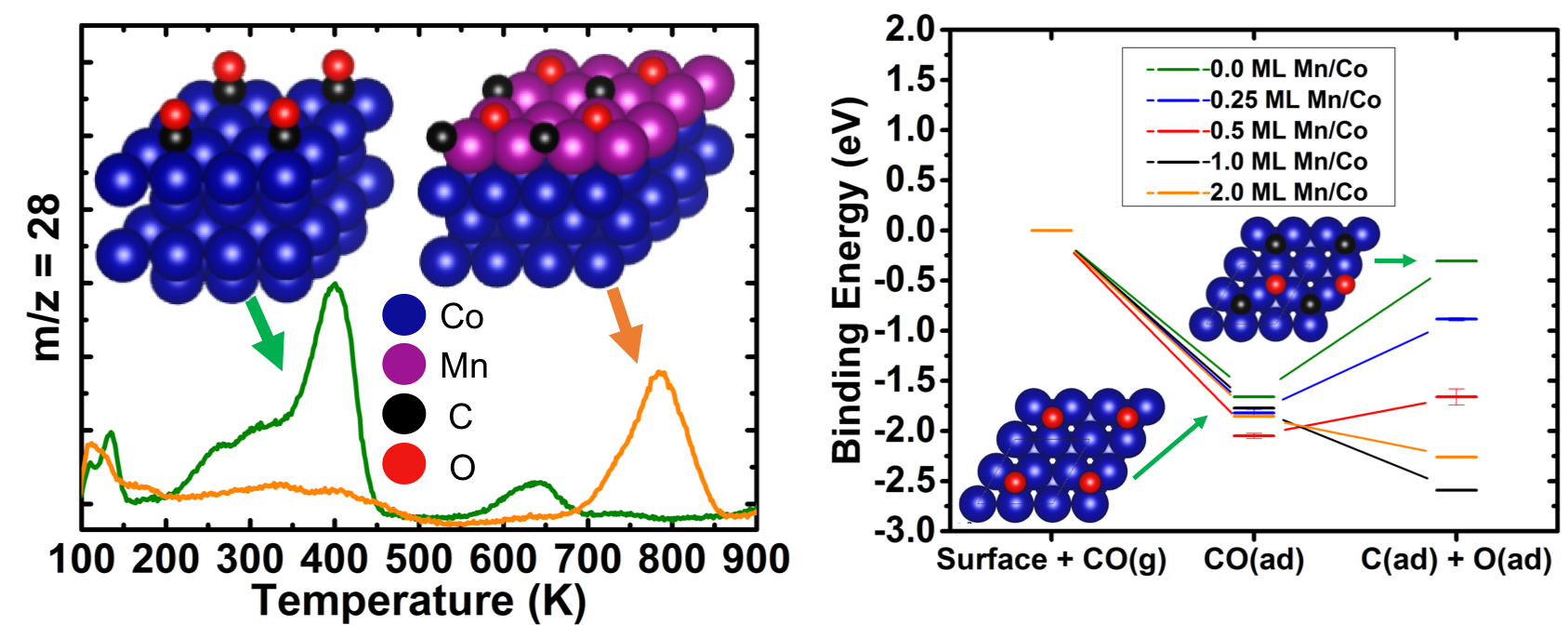
Our group collaborates with Prof. C. Buddie Mullins at UT Austin to study surface reactions related to the Fischer-Tropsch process. Using density functional theory, we studied Mn/Co models for the dissociation of CO. In agreement with experiments, we saw that Mn coverages on Co promoted the adsorption and dissociation of CO.
Selective Oxidation of Acetaldehyde to Acetic Acid on Pd-Au Bimetallic Model Catalysts
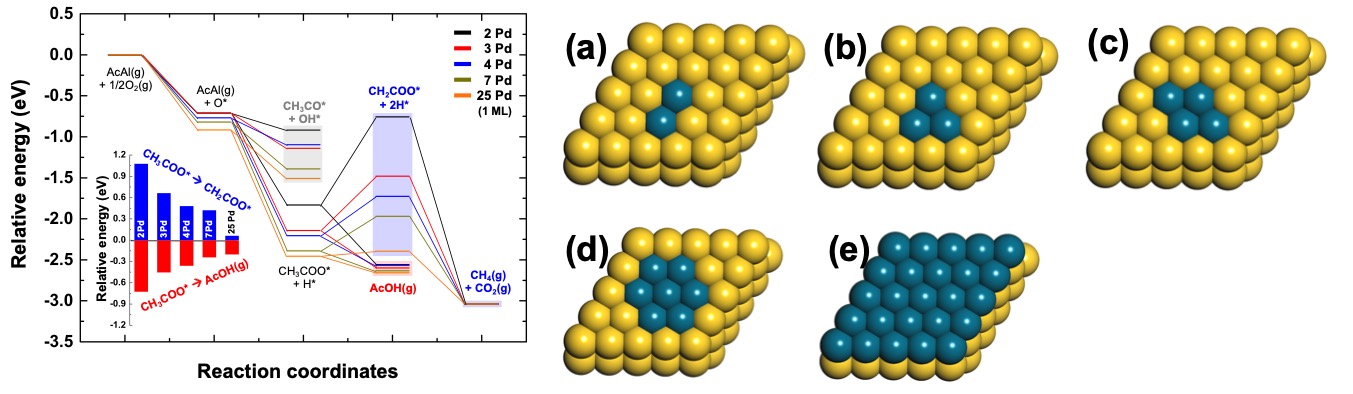
Acetic acid is a widely employed reactant in the chemical industry, and it is also used as a food ingredient. Here, we reported a catalytic reaction pathway for the gas-phase selective oxidation of acetaldehyde to acetic acid on a Pd-Au(111) heterogeneous moedel catalyst with different Pd coverages. Different size of Pd ensembles could change the binding energy of intermediates, and also affected the way of adsorption. We found that stabilization of CH2COO* as size of Pd ensemble increases, which can be directly connected to selectivity. This work is collaborated with Prof. C. Buddie Mullins at UT Austin.
References
M. Jing, W. Song, Y. Li, Z. Zhao, J. Liu, and G. Henkelman,
Theoretical study of structure sensitivity on Au doped CeO2 surfaces for formaldehyde oxidation: the effect of crystal planes and Au doping,
Chem. Eng. J. 433, 133599 (2022).
Z. Duan and G. Henkelman,
Atomic-scale Mechanisms of Electrochemical Pt Dissolution,
ACS Catal. 11, 14439-14447 (2021).
H. Zheng, H. Li, L. Luo, Z. Zhao, and G. Henkelman,
Factors that Influence Hydrogen Binding at Metal-Atop Sites,
J. Chem. Phys. 155, 024703 (2021).
Z. Duan and G. Henkelman,
Identification of Active Sites of Pure and Nitrogen-Doped Carbon Materials for Oxygen Reduction Reaction Using Constant-Potential Calculations,
J. Phys. Chem. C 124, 12016-12023 (2020).
S. Han, K. Shin, G. Henkelman, C. B. Mullins,
Selective Oxidation of Acetaldehyde to Acetic acid on Pd–Au Bimetallic Model Catalysts,
ACS Catal. 9, 4360-4368 (2019).
L. Xu, D. Mei, and G. Henkelman,
Adaptive kinetic Monte Carlo simulation of methanol decomposition on Cu(100)
J. Chem. Phys. 131, 244520 (2009).
R. A. Ojifinni, J. Gong, N. S. Froemming, D. Flaherty, M. Pan, G. Henkelman, and C. B. Mullins,
Carbonate formation and decomposition on atomic oxygen precovered Au(111)
J. Am. Chem. Soc. 130, 11250 (2008).
R. A. Ojifinni, N. S. Froemming, J. Gong, M. Pan, T. S. Kim, J. M. White, G. Henkelman, and C. B. Mullins,
Water-enhanced low-temperature CO oxidation and isotope effects on atomic oxygen covered Au(111)
J. Am. Chem. Soc. 130, 6801 (2008).
G. Henkelman, A. Arnaldsson, and H. Jónsson,
Theoretical calculations of CH4 and H2 associative desorption from Ni(111): Could subsurface hydrogen play an important role?,
J. Chem. Phys. 124, 044706 (2006).
G. Henkelman and H. Jónsson,
Theoretical calculations of dissociative adsorption of CH4 on an Ir(111) Surface,
Phys. Rev. Lett. 86, 664 (2001).